Sindrome di Ehlers-Danlos
Sinonimi

EDS, sindrome di Ehlers-Danlos-Meekeren, sindrome di Van Meekeren, Fibrodysplasia elastica generalisata, Dermatolysis, Cutis hyperelastica, "rubber skin", ecc.
Francese: Laxité articulaire congénitale multiple
Engl .: sindrome di Danlos, sindrome di Meekeren-Ehlers-Danlos, sindrome di Chernogubov, sindrome di Sack, sindrome di Sack-Barabas, sindrome di Van Meekeren I.
Russo: sindrome di Chernogubov
Definizione / introduzione
Il Sindrome di Ehlers-Danlos (EDS) riassume un gruppo eterogeneo, genetico Malattie del tessuto connettivo insieme, causati da disturbi nella sintesi del collagene, una proteina strutturale del Tessuto connettivo, sintomi condizionali e caratteristici di pelle, Giunti e organi interni mostre.
frequenza
Il Sindrome di Ehlers-Danlos è raro. La prevalenza nella popolazione totale è di 1: 5000; Il 90% di loro è tra questi Tipi I, II e III affetti (30% ciascuno) e circa il 10% di tipo IV Le altre forme sono raramente osservate.
Il Tipi I-III diventare dominanza autosomica ereditato, ad es. ci deve essere solo un gene difettoso perché la malattia scoppi. Gli altri ragazzi lo faranno autosomica recessivacioè devono esserci due geni difettosi, o X-linkedcioè Trasmissione del cromosoma sessuale, ereditata.
storia
questo è stato descritto per la prima volta Sindrome di Ehlers-Danlos nell'anno 1668 a partire dal Job Janszoon di Meekeren (1611-1666), chirurgo di Amsterdam. Aveva scoperto il sintomo di un'anormale elasticità in uno spagnolo che poteva tirare la pelle del mento fino agli occhi e sopra il petto. Tuttavia, non ha osservato altre anomalie.
Primo 1891 ha creato il dermatologo Chernogubov una descrizione completa del quadro clinico compreso il coinvolgimento articolare e vascolare, motivo per cui nella medicina russa
Letteratura tecnica fino ad oggi il nome "Sindrome di Chernogubov" è comune.
Seguirono ulteriori descrizioni 1901 dal dermatologo danese Eduard Ehlers (1863-1937) e 1908 dal dermatologo di Parigi Henri A. Danlos (1844-1912). Non è stato fino al 1933 che "Sindrome di Ehlers-Danlos" come il nome della malattia prevale.
1949 c'erano le prime intuizioni sulla frequenza familiare della malattia e 1972 era un errore genetico correlato a questo Sindrome di Ehlers-Danlos scoperto. Nel 1986 è stata stabilita una classificazione preliminare in 10 tipi, che è stata modificata nel 1997 in una versione semplificata con la suddivisione in sei tipi principali.
cause
La causa della malattia è a Difetto genetico. C'è un cambiamento (mutazione) nei geni che compongono la proteina strutturale collagene descrivere sul genoma umano, il DNA. La mutazione porta ad una alterata struttura e / o ridotta sintesi del collagene, che porta ad una ridotta resistenza dell'intero tessuto connettivo.
Tutti e due Tipi I e II è una mutazione nel gene collagene V, in cui Tipi IV una mutazione in Collagene III.
Sintomi
A causa della sintesi disturbata e ridotta del collagene, il Sindrome di Ehlers-Danlos le parti del corpo particolarmente ricche di tessuto connettivo: i pelle, Giunti e Vasi sanguigni. Poiché il tessuto connettivo manca di forza, è troppo allungabile e si strappa molto rapidamente, il che è particolarmente vero per i vasi sanguigni troppo piccoli, ma a volte troppo sanguinamento massiccio può condurre. Una complicazione importante è la formazione di rigonfiamenti nei vasi, i cosiddetti. aneurismi, con il rischio di rottura.
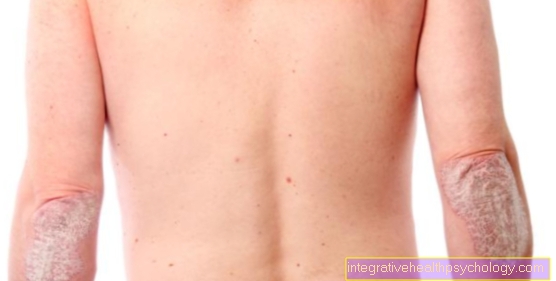
Il Sintomo principale della pelle è il più pronunciato Cutis iperelasticosul lato del collo, al di sopra di Giunti e anche in viso può essere sollevato fino a 4 cm o più. Dopo averlo lasciato, torna immediatamente alla posizione di partenza, motivo per cui ha preso il nome "Pelle di gomma" indossare. In generale, la pelle è notevolmente sottile (come la carta da sigarette), morbida e vellutata ("Marshmallow Skin").
Le ferite mostrano una guarigione ritardata della ferita, quindi le suture impiegano da 3 a 4 volte più tempo per guarire. Atrofici o ipertrofici, quelli inferiori spesso si sviluppano dalle cuciture cicatrice. Inoltre, c'è la formazione di rigonfiamenti cutanei pieni di liquido (succulenti) (pseudotumori mollusoidi) su aree del corpo fortemente stressate, come Articolazione del ginocchio e del gomito, alla formazione di cavigliere ("Paranocche") sopra Dorso della mano e del piede e di noduli sul tacco.
Il Le articolazioni possono essere allungate eccessivamente (HYPERFlessibilità), spesso mobili in direzioni indesiderate e privi di forza a causa di legamenti articolari allentati (Lassità dei legamenti). Ciò può causare movimenti insoliti, come quelli da cui ci si potrebbe aspettare "contorsionisti" conosce. Le articolazioni tendono a contorsioni (dislocazioni) e malposizioni. Quelli sono particolarmente colpiti spalla- e Articolazioni della caviglia, il Rotula (patella), il Articolazione temporo-mandibolare (Articolazione temporo-mandibolare) e più raramente quello Articolazione del gomito. La documentazione dell'eccessiva mobilità delle articolazioni (ipermobilità) viene eseguita dal Il punteggio di Beightonche conferma l'ipermobilità su 5 punti su 9 possibili.
Altri sintomi delle articolazioni sono problemi articolari generalizzati, dolore al collo cronico, mossa- e Dolore all'anca, Giunto e Dolori muscolaridifficili da trattare. A volte punti dolenti ("Tender ponits"), definite come un'area che reagisce dolorosamente a carichi di pressione pari o inferiori a 4 kg. Inoltre, vi è un aumento del rischio di fratture a causa della ridotta massa ossea associata a una struttura ossea anormale.
A causa della fragilità del tessuto connettivo dei vasi sanguigni, c'è una pronunciata tendenza a ematomi, spontaneamente o come risultato di un trauma,
principalmente nelle aree a rischio di lesioni. Questo è seguito da un tipico nelle aree colpite pigmentazione marrone.
Dopo gli infortuni si osserva una tendenza a sanguinamento prolungato con valori di coagulazione normali. La fragilità dei vasi sanguigni più grandi può essere innescata da sforzo, incidenti, gravidanza o nascita portare a emorragie massicce e pericolose per la vita.
Poiché anche altre strutture del tessuto connettivo sono inferiori, può diventarlo viscere (Ernia / ernia inguinale)), curvatura della colonna vertebrale (Scoliosi), Crepe (rottura) del intestino e il utero (Utero), sacche (Aneurisma) dei vasi sanguigni e costrizione dei polmoni dall'aria libera nel torace (pneumotorace) venire.
In rari casi, i cambiamenti agli occhi sono associati a EDS come Astigmatismo (astigmatismo) o Stella verde (glaucoma) osservare.
diagnosi
La diagnosi viene effettuata sulla base dell'aspetto clinico, dei sintomi ed è integrata da un esame familiare (storia familiare). Inoltre, a Biopsia cutanea in cui il tessuto cutaneo rimosso viene esaminato utilizzando un microscopio elettronico e viene valutata la sua struttura di collagene. La differenziazione nei diversi tipi di Sindrome di Ehlers-Danlos avviene mediante analisi di sequenza del DNA.
Classificazione / tipi
Tipo I, II: Ragazzo classico; Eredità: autosomica dominante; Sintomi principali: Iperelasticità e fragilità della pelle, cicatrici atrofiche, ipermobilità articolare; Causa: disturbo della formazione del collagene V.
Tipo III: Tipo ipermobile; Eredità: autosomica dominante; Sintomi principali: ipermobilità articolare generalizzata, coinvolgimento della pelle (iperelasticità e / o pelle morbida e vulnerabile); Causa: disturbo della formazione del collagene V.
Maggiori informazioni su questo tipo importante su: Sindrome di Ehlers-Danlos, tipo III
Tipo IV: Tipo vascolare; Eredità: autosomica dominante; Sintomi principali: pelle sottile e traslucida, rotture delle arterie, dell'intestino e dell'utero, tendenza pronunciata all'ematoma; Causa: disturbo della formazione di collagene III
Tipo V: corrisponde al tipo I.
Tipo VI: Tipo cifoscoliotico; Eredità: autosomica recessiva; Sintomi principali: diminuzione della tensione del Muscolatura già alla nascita ("Floppy infant"), sviluppo ritardato di trattenere e sostenere riflessi, flessione laterale del Colonna vertebrale (Scoliosi), Causa ultima: Deficit di lsysl idrossilasi
Tipo VII A / B: Tipo artrocalastico; Eredità: autosomica dominante; Sintomi principali: grave ipermobilità generalizzata delle articolazioni con lussazioni ripetute, lussazione bilaterale congenita dell'anca; Causa ultima: Disturbo del collagene di tipo I.
Tipo VII C: Tipo dermatosparattico; Eredità: autosomica dominante; Sintomi principali: fragilità cutanea pronunciata, rilassamento cutaneo, Causa ultima: Carenza di procollagene I peptidasi N-terminale
Terapia e profilassi
Neanche uno causale ancora un terapia sintomatica è attualmente possibile, quindi la profilassi dei danni consequenziali è in primo piano. Si dovrebbero evitare lesioni e un maggiore stress sulle articolazioni. Quindi dovrebbe certo gli sportche sono associati ad un aumentato rischio di lesioni non vengono esercitati. A causa dell'aumentato rischio di complicanze gravidanza e nascita tutti e due Tipi I, II, IV e VI è richiesto uno stretto monitoraggio.
Dovrebbe essere usato anche con il raffreddore terapia antitosse e generalmente al Regolazione della consistenza delle feci da rispettare, esiste una cosa del genere Rottura del colon (Rottura del colon) e a pneumotorace può essere evitato. Attraverso la fisioterapia precoce, soprattutto nei bambini, è possibile stabilizzare le articolazioni ipertrattenibili, il che porta ad alleviare i sintomi dell'intero sistema muscolo-scheletrico.
ferite è necessario prestare particolare attenzione e le operazioni devono essere eseguite solo in casi di emergenza perché la guarigione della ferita richiede da 3 a 4 volte più lentamente del solito.
previsione
Paziente con Sindrome di Ehlers-Danlos di solito hanno un'aspettativa di vita normale. Tuttavia, la malattia è progressiva, quindi porta a un deterioramento sempre maggiore della salute. Le ferite della pelle e le lussazioni articolari influiscono sulla qualità della vita del paziente, mentre la rottura dei grandi vasi può essere pericolosa per la vita.
Aspettativa di vita
La sindrome di Ehlers-Danlos è una malattia cronica per la quale non esiste ancora un trattamento causale e quindi nessuna cura. Ciò significa che, dato lo stato attuale della tecnologia medica, non c'è modo di fare nulla per le cause della sindrome di Ehlers-Danlos e di curarla completamente. Sfortunatamente, non si è ancora in grado di combattere e curare i sintomi che si verificano. L'unica cosa che puoi fare è incoraggiare il paziente affetto a stare sempre attento a non sottoporre a sforzi eccessivi le articolazioni e ad evitare lesioni alla pelle quando possibile. Gli interventi chirurgici dovrebbero essere eseguiti solo in caso di emergenza e se non ci sono alternative adeguate.
Nella maggior parte dei casi è progressivo con un peggioramento crescente dei sintomi e delle menomazioni nella vita quotidiana del paziente. A seconda del tipo di malattia, la malattia ha effetti diversi sulla vita delle persone colpite. I cambiamenti nelle articolazioni a volte portano all'osteoartrosi e all'artrite nella prima infanzia, in modo che i bambini imparino in seguito a camminare ei loro piedi possano deformarsi. Con l'aumento del rischio di distacco di retina o emorragia retinica, anche la vista è compromessa.
La forza con cui i sintomi sono alla fine pronunciati in ogni individuo dipende fortemente dal tipo di sindrome di Ehlers-Danlos e possono anche variare notevolmente all'interno dei singoli sottotipi. Per la maggior parte dei tipi di sindrome di Ehlers-Danlos, l'aspettativa di vita è normale. Nei pazienti con sindrome di Ehlers-Danlos di tipo IV, che colpisce i vasi, l'aspettativa di vita è significativamente ridotta a causa di gravi complicazioni, come il rischio di rottura spontanea di un'arteria, in particolare dell'arteria principale (lacerazione aortica med.) O dell'intestino crasso.
È di circa 37 anni per le donne e 34 anni per gli uomini.
Con la sindrome di Ehlers-Danlos di tipo VI si può anche ipotizzare una ridotta aspettativa di vita.