Sindrome di Marfan
Sinonimi in senso lato
Fibrillopatia di tipo 1; Sindrome di Aracnodattilia; Dito di ragno; Sindrome di Achard-Marfan; MFS
definizione
La sindrome di Marfan è una rara malattia genetica del Tessuto connettivo con cambiamenti anormali cuore, Navi, occhio e scheletro con il sintomo principale di arti lunghi, stretti o di ragno. La sindrome di Marfan si basa su un cambiamento genetico (mutazione) del gene della fibrillina-1, che può essere ereditato in modo autosomico dominante dai genitori o occasionalmente presentarsi come una nuova mutazione.
Epidemiologia
Con una prevalenza (Incidenza della malattia) a partire dal 1:10.000 e ca. 8000 le persone colpite in Germania possono Sindrome di Marfan essere annoverato tra le malattie rare. Uomini e donne sono ugualmente colpiti perché la malattia non colpisce i cromosomi sessuali. La sindrome di Marfan ha preso il nome da una delle sue prime descrizioni, Antoine Marfanche ha osservato dita anormalmente lunghe e strette in una bambina nel 1896.
Il tessuto connettivo
Il tessuto connettivo del corpo umano si trova al di fuori delle cellule, si parla anche della matrice extracellulare che si trova ovunque nel corpo. Puoi trovarlo principalmente in osso, cartilagine, tendini e Vasi sanguigni, ma anche in tutti gli altri sistemi di organi, il tessuto connettivo ha il compito di legare, avvolgere e servire da struttura guida. Il tessuto connettivo è composto da proteine (aminoacidi) e catene di zucchero (saccaridi). Queste proteine possono unirsi con le catene di zucchero per formare grandi aggregati e quindi formare ad es. le fibrille di collagene (elementi costitutivi delle fibre di collagene) del osso o il capelli. Le microfibrille del tessuto connettivo sono anche aggregati costituiti da componenti fibrillinici, altre proteine e zuccheri. Queste microfibrille si trovano nella maggior parte dei tessuti connettivi e si trovano sempre sulla superficie delle fibre elastiche, che ad es. i tessuti del pelle danno la loro elasticità. Ma hanno anche i loro compiti nelle fibre della cartilagine e dei tendini. Per ciascuna delle diverse fibrilline che costituiscono la spina dorsale delle microfibrille, c'è l'informazione genetica necessaria per produrle proteine è necessario su diversi cromosomi.
I compiti della fibrillina-1 includono la formazione di microfibrille, ma questo deve essere fatto durante la sua produzione (sintesi) nella cella (fibroblasti) sono stati piegati correttamente, ovvero hanno adottato la struttura corretta. Quando si formano le microfibrille, la fibrillina necessita di varie molecole di supporto che sono necessarie per la stabilizzazione quando le fibrille sono reticolate. Questo include anche quello calcio.
Quindi è comprensibile che la mancanza di microfibrille funzionali porti ad un'espansione dell'arteria principale (Dilatazione aortica), poiché la loro funzione stabilizzante non è più disponibile a causa del difetto della fibrillina.
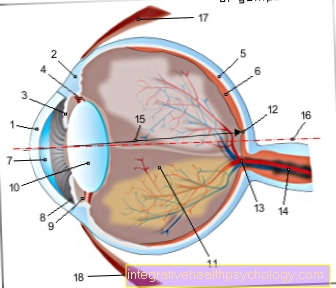
L'eccessiva crescita degli arti è dovuta alla mancanza di una funzione regolatrice delle microfibrille nella crescita longitudinale delle ossa lunghe. Lo stesso vale per le fibre zonulari dell'occhio (l'apparato di sospensione del Obiettivo dell'occhio) che perdono immensamente stabilità e diventano così "Shrapnel di lenticchie"per condurre.
Per poter effettuare questa necessaria corretta piegatura delle fibrille e della loro formazione aggregata, esse devono essere calcio essere vincolati, il che li protegge anche dal degrado prematuro. Spesso, tuttavia, è proprio la regione della fibrillina interessata dalla mutazione responsabile del legame del calcio. Di conseguenza, la piegatura può quindi fallire e le microfibrille sono soggette a degradazione accelerata.
In sintesi, i vari sintomi della sindrome di Marfan sono causati da microfibrille indebolite o addirittura mancanti che non possono essere costruite correttamente a causa di un gene della fibrillina difettoso.
terapia

Primo passo nella terapia del Sindrome di Marfan di solito è un adattamento immediato dello stile di vita dopo la diagnosi. Lesioni gravi come a Colpo di frusta (Trauma da accelerazione) o si scontra con altri, come hanno fatto quando pallacanestro, pallavolo o Calcio dovrebbe essere evitato se possibile con la sindrome di Marfan esistente, in quanto ciò crea il rischio di scindere il aorta (Dissezione) può essere aumentata. Questo è problematico proprio perché molti pazienti con sindrome di Marfan hanno scelto sport come il basket a causa della loro altezza. Lo stesso pericolo si riscontra negli sport in cui valori di pressione sanguigna massimamente elevati (Picchi di pressione sanguigna) si verificano, come ad es. al bodybuilding il caso è. In ogni caso è essenziale un monitoraggio regolare delle condizioni dei vasi colpiti Sindrome di Marfan visualizzato. Se il diametro aortico è inferiore a 40 millimetri, è sufficiente un controllo annuale. Se c'è il rischio di rottura dell'aorta, la chirurgia è inevitabile.
Se c'è un desiderio urgente di avere figli, una storia familiare con scissione dell'aorta o allo stesso tempo chiusura inadeguata delle valvole cardiache (Rigurgito aortico o mitralico) un'operazione può già essere eseguita con un'espansione dell'aorta (Aneurisma) di 50 millimetri. La mortalità della procedura aumenta dall'1% per le operazioni pianificate al 27% per le operazioni di emergenza.
Di solito la tecnica consiste nell'estrarre la parte ingrandita dell'aorta (Chirurgia dopo Bentall). Qui l'aorta ascendente e il Valvola aortica attraverso un lembo portante "Composito“Protesi sostituita. Uno svantaggio di questa procedura è l'assunzione per tutta la vita di anticoagulanti (anticoagulanti). Per evitare ciò, viene sempre più utilizzata la tecnologia di conservazione della valvola David o Yacoub usato, sostituendo solo l'aorta con una protesi. Tuttavia, la valvola conservata spesso degenera negli anni, il che rende necessaria una seconda operazione. Se, d'altra parte, c'è una debolezza della valvola cardiaca sinistra (Rigurgito della valvola mitrale) questo dovrebbe essere sostituito da una protesi valvolare per evitare un'ulteriore espansione dell'aorta ascendente. Anche qui diventa inevitabile un'inibizione permanente della coagulazione del sangue. Se la valvola mitrale viene ricostruita, questi farmaci possono essere eliminati, il che è particolarmente vantaggioso per i pazienti giovani.
Il trattamento della dislocazione della lente di solito consiste nella rimozione chirurgica della lente e nella fornitura di una struttura di occhiali corrispondentemente altamente rifrattiva o nella rimozione delle fibre della lente eccessivamente lunghe preservando la lente e la vista. Spesso, tuttavia, la miopia può essere corretta anche regolando a bicchieri o lenti a contatto sufficiente.
Nell'area del sistema scheletrico, la scoliosi della colonna vertebrale, che si verifica nella metà dei pazienti, può essere utile, specialmente nei bambini, quando indossano un corsetto. L'obiettivo è prevenire il deterioramento della colonna vertebrale piegata durante la crescita. Tuttavia, non causa un raddrizzamento permanente. Se la scoliosi supera i 40 gradi, un chirurgo ortopedico dovrebbe prendere in considerazione il raddrizzamento chirurgico della colonna vertebrale per prevenire problemi polmonari e mal di schiena.
Nella maggior parte dei casi, il trattamento dell'imbuto o del torace della chiglia viene eseguito solo per motivi estetici; solo nei casi più rari si ha compressione dei polmoni, del cuore o dell'aorta, che deve essere corretta chirurgicamente.
Il disagio in caso di piedi piatti può passare solette e calzature adeguate alleviano il dolore e migliorano il comfort della deambulazione. Il restauro chirurgico dell'arco del piede è necessario solo raramente nella sindrome di Marfan.
Il rigonfiamento dell'acetabolo è dovuto solo a circa il 5% degli adulti affetti Dolore all'anca e la mobilità ridotta richiede un trattamento e viene fornito con a articolazione artificiale dell'anca trattata.
profilassi
L'uso profilattico di farmaci per abbassare la pressione sanguigna den Beta bloccanti, per ritardare l'espansione dell'aorta, è particolarmente evidente nei pazienti più giovani, non operati con Sindrome di Marfan altrettanto efficace. Non è stato possibile stabilire l'efficacia in pazienti anziani o già trattati.
La profilassi dell'endocardite, ovvero la terapia antibiotica per evitare l'infiammazione del rivestimento interno del cuore, deve essere eseguita nei pazienti Marfan per tutti gli interventi o lesioni gravi, poiché sono particolarmente sensibili a questo a causa dei vasi e delle valvole cardiache danneggiati.
previsione
Il Aspettativa di vita per la sindrome di Marfan è significativamente ridotto nei pazienti non trattati. Con una terapia ottimale, invece, si possono raggiungere fino a 60 anni di vita, a condizione che la malattia e le sue complicanze potenzialmente letali vengano diagnosticate in una fase precoce. Tuttavia, la valutazione prognostica nei pazienti che non soddisfano tutti i criteri per una malattia è problematica. Dal momento che Sindrome di Marfan è un continuum fenotipico che può variare dalla sindrome neonatale di Marfan con aspettativa di vita inferiore a un anno a forme lievi senza manifestazioni esterne e quasi nessuna complicanza nell'area cardiovascolare, spesso è difficile stimarne il decorso nel tempo.